EN
Oxygen, as one of the most common and important elements in nature, seems ordinary but solid oxygen is actually a very unique crystal. Among many simple diatomic molecule systems, oxygen is the only one with magnetic properties, and solid oxygen is the only elemental solid that is an antiferromagnetic insulator at low temperatures. Theoretical studies suggest that pressure can cause the magnetic properties of oxygen molecules to disappear, but no structural-experimental data has supported or proven this phenomenon until now. Recently, a team led by Dr. Federico Gorelli and Dr. Philip Dalladay-Simpson from HPSTAR using single crystal X-ray diffraction to reveal the structural origin of the disappearance of oxygen's magnetic properties. Single crystal X-ray diffraction showed that at 18.1 GPa, the epsilon phase undergoes a structural phase transition, changing from a magnetic spin liquid state to a non-magnetic state. The related results were published in the recent issue of Physical Review Letters and the paper is selected for a highlight story by the editor of Physics Magazine of APS.
Oxygen is unique among simple diatomic molecules for its molecular spin S = 1, giving rise to additional magnetic interactions and a remarkably rich phase diagram [Fig. 1(b)]. Below 8–10 GPa, intermolecular spin–spin interactions stabilize the antiferromagnetic (AFM) α and δ phases—still the only known insulating elemental AFM phases. Above 10 GPa, at room temperature, oxygen transforms into the ε–O₂ phase: a deep-red semiconductor composed of distinctive (O₂)₄ tetramer clusters [1]. Neutron diffraction has shown that the δ–ε transition suppresses long-range AFM order [2], yet the ultimate fate of the S = 1 spins has remained an open question.
A seminal theoretical study [3] proposed that ε–O₂ actually comprises two sub-phases, ε₁ and ε₀, separated by a first-order transition. In ε₁ (< 20 GPa), (O₂)₄ tetramers preserve S = 1 spins with short-range AFM correlations. In ε₀ (> 20 GPa), the spins collapse to S = 0. Tosatti and co-workers described ε–O₂ as a remarkable case of spin physics: contrary to early assumptions, first-principles calculations indicate that spins survive in ε₁. Here, O₂ molecules magneto-elastically self-assemble into “quartets” to minimize AFM coupling energy; these quartets resonate into singlets, erasing long-range AFM order but retaining intra-cluster spin activity—an exotic “spin-1 quantum liquid” or “mini-droplet.” While quantum spin liquids are well known in S = 1/2 systems (e.g., Bethe’s 1D chain, Anderson’s 2D RVB), the ε₁ state is a rare solid-state example for S = 1. The ε₁–ε₀ transition is predicted to coincide with spin collapse and to explain the long-standing minimum in the IR stretching frequency versus pressure [4].
The team, exploiting the high performance of the ID27 beamline at the ESRF–EBS synchrotron, achieved high-precision single-crystal refinements of ε–O₂ in helium. Their results reveal distinct discontinuities at 18.1 ± 0.5 GPa in the pressure evolution of the O–O distance and one rhomboid diagonal. These structural changes are mirrored in the lattice parameters and volume–pressure relations: parameters β and a follow separate compression trends below and above 18.1 GPa, while the unit-cell volume is best described by two distinct Birch–Murnaghan equations of state. This behavior confirms a first-order isostructural transition—precisely as predicted for the ε₁–ε₀ spin-collapse boundary [3]. As Dr. Gorelli notes, “This discovery allows us to redefine the phase diagram and explain spectroscopic anomalies, such as the IR-frequency minimum[4]unresolved for a quarter of a century- the short range antiferromagnetic coupling between two neighbour O2 molecules decreases the IR stretching frequency with pressure. When this tendency changes and starts to increase after the minimum it is an indication that O2 molecules have lost the individual spin S=1 and behave as “standard” molecules which generally show a hardening of the stretching frequency with increasing pressure. The short range antiferromagnetic coupling between two neighbour O2 molecules decreases the IR stretching frequency with pressure. When this tendency changes and starts to increase after the minimum it is an indication that O2 molecules have lost the individual spin S=1 and behave as “standard” molecules which generally show a hardening of the stretching frequency with increasing pressure.”
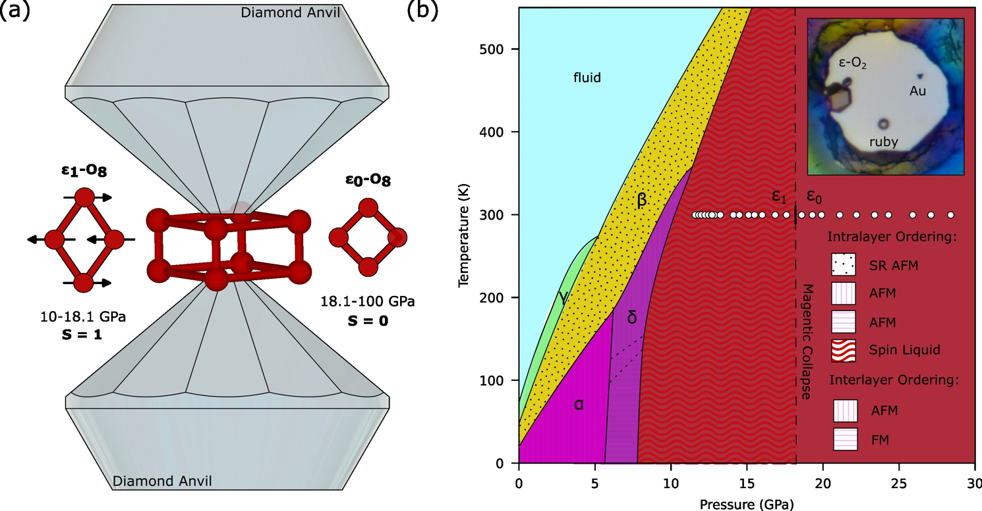
Figure 1: (a) Illustration of the O8 molecular unit in the spin liquid, S=1, (left of diamonds) and spinless states, S=0, (right of diamonds). (b) Structural and magnetic phase diagram of oxygen, empty circles represent single crystal refinements and the dashed line the evidenced spin collapse of the oxygen molecule. The inset is a high-quality annealed single crystal grown in a helium environment.
References
[1] L. F. Lundegaard et al., Nature (London) 443, 201 (2006); H. Fujihisa et al., Phys. Rev. Lett. 97, 085503 (2006).
[2] I. N. Goncharenko, Phys. Rev. Lett. 94, 205701 (2005).
[3] Y. Crespo et al., Proc. Natl. Acad. Sci. U.S.A. 111, 10427 (2014); earlier suggestions by H.V. Gomonay, V. M. Loktev, Phys. Rev. B 76, 094423 (2007); M. Bartolomei et al, Phys. Rev. B 84, 092105 (2011).
[4] F.A. Gorelli et al Phys. Rev. Lett., 83, 4093 (1999).
生命之源-氧,作为自然界中最重要且分布最广泛的元素之一,看似寻常,实则非常独特:在众多简单双原子分子体系中,氧是唯一具有磁性的,其固体形态也是唯一在低温下呈现反铁磁绝缘性的单质。理论研究预测压力可导致氧分子磁性消失,但这一现象的结构性实验证据一直缺失。近日,北京高压科学研究中心的Federico Gorelli研究员与Philip Dalladay-Simpson研究员领衔的团队,通过高压原位单晶X射线衍射测量,首次揭示了氧分子磁性消失的结构起源。研究表明,在18.1 GPa压力下,氧的ε相发生等结构相变,与氧分子从磁性自旋液态转变为非磁态的压力一致。相关成果发表于近期的Physical Review Letters。该工作被美国物理学会《物理》杂志的编辑选为亮点文章报道。

自由 Freedom
合作 Collaboration
顶尖 Excellence